AutoDock Vina Manual
Contents
- Features
- License
- Tutorial
- Frequently Asked Questions
- Platform Notes and Installation
- Other Software
- Usage
- Virtual Screening
- History
- Citation
- Getting Help
- Reporting Bugs
Features
| AccuracyAutoDock Vina significantly improves the average accuracy of the binding mode predictions compared to AutoDock 4, judging by our tests on the training set used in AutoDock 4 development.[*]
Additionally and independently, AutoDock Vina has been tested against a virtual screening benchmark called the Directory of Useful Decoys by the Watowich group, and was found to be “a strong competitor against the other programs, and at the top of the pack in many cases”. It should be noted that all six of the other docking programs, to which it was compared, are distributed commercially. AutoDock Tools Compatibility For its input and output, Vina uses the same PDBQT molecular structure file format used by AutoDock. PDBQT files can be generated (interactively or in batch mode) and viewed using MGLTools. Other files, such as the AutoDock and AutoGrid parameter files (GPF, DPF) and grid map files are not needed. |
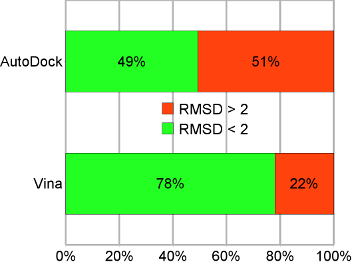 Binding mode prediction accuracy on the test set. “AutoDock” refers to AutoDock 4, and “Vina” to AutoDock Vina 1. |
Ease of Use
Vina’s design philosophy is not to require the user to understand its implementation details, tweak obscure search parameters, cluster results or know advanced algebra (quaternions). All that is required is the structures of the molecules being docked and the specification of the search space including the binding site. Calculating grid maps and assigning atom charges is not needed. The usage summary can be printed with “vina --help“. The summary automatically remains in sync with the possible usage scenarios.
Implementation Quality
- By design, the results should not have a statistical bias related to the conformation of the input structure.
- Attention is paid to checking the syntactic correctness of the input and reporting errors to the user in a lucid manner.
- The invariance of the covalent bond lengths is automatically verified in the output structures.
- Vina avoids imposing artificial restrictions, such as the number of atoms in the input, the number of torsions, the size of the search space, the exhaustiveness of the search, etc.
Flexible Side Chains
Like in AutoDock 4, some receptor side chains can be chosen to be treated as flexible during docking.
| SpeedAutoDock Vina tends to be faster than AutoDock 4 by orders of magnitude.[*]
Multiple CPUs/Cores Additionally, Vina can take advantage of multiple CPUs or CPU cores on your system to significantly shorten its running time. World Community Grid Qualified projects can run AutoDock Vina calculations for free on the massively parallel World Community Grid. Existing projects using AutoDock Vina there include those targeting AIDS, Malaria, Leishmaniasis and Schistosomiasis. Some of these projects average over 50 years worth of computation per day. |
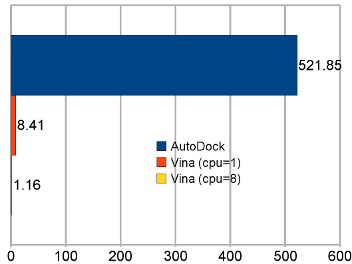 Average time per receptor-ligand pair on the test set. “AutoDock” refers to AutoDock 4, and “Vina” to AutoDock Vina 1. |
License
AutoDock Vina is released under a very permissive Apache license, with few restrictions on commercial or non-commercial use, or on the derivative works. The text of the license can be found here.
Tutorial
If you have never used AutoDock Vina before, please study the Video Tutorial before attempting to use it.
 |
This video tutorial demonstrates molecular docking of imatinib using Vina with AutoDock Tools and PyMOL |
Frequently Asked Questions
How to get started learning to use Vina?
Watching the video tutorial might be the best way to do that.
What is the meaning or significance of the name “Vina”? Why was it developed?
Please see this mailing list post.
How accurate is AutoDock Vina?
See Features
It should be noted that the predictive accuracy varies a lot depending on the target, so it makes sense to evaluate AutoDock Vina against your particular target first, if you have known actives, or a bound native ligand structure, before ordering compounds. While evaluating any docking engine in a retrospective virtual screen, it might make sense to select decoys of similar size, and perhaps other physical characteristics, to your known actives.
What is the difference between AutoDock Vina and AutoDock 4?
AutoDock 4 (and previous versions) and AutoDock Vina were both developed in the Molecular Graphics Lab at The Scripps Research Institute. AutoDock Vina inherits some of the ideas and approaches of AutoDock 4, such as treating docking as a stochastic global opimization of the scoring function, precalculating grid maps (Vina does that internally), and some other implementation tricks, such as precalculating the interaction between every atom type pair at every distance. It also uses the same type of structure format (PDBQT) for maximum compatibility with auxiliary software.
However, the source code, the scoring funcion and the actual algorithms used are brand new, so it’s more correct to think of AutoDock Vina as a new “generation” rather than “version” of AutoDock. The performance was compared in the original publication [*], and on average, AutoDock Vina did considerably better, both in speed and accuracy. However, for any given target, either program may provide a better result, even though AutoDock Vina is more likely to do so. This is due to the fact that the scoring functions are different, and both are inexact.
What is the difference between AutoDock Vina and AutoDock Tools?
AutoDock Tools is a module within the MGL Tools software package specifically for generating input (PDBQT files) for AutoDock or Vina. It can also be used for viewing the results.
Can I dock two proteins with AutoDock Vina?
You might be able to do that, but AutoDock Vina is designed only for receptor-ligand docking. There are better programs for protein-protein docking.
Will Vina run on my 64-bit machine?
Yes. By design, modern 64-bit machines can run 32-bit binaries natively.
Why do I get “can not open conf.txt” error? The file exists!
Oftentimes, file browsers hide the file extension, so while you think you have a file “conf.txt“, it’s actually called “conf.txt.txt“. This setting can be changed in the control panel or system preferences.
You should also make sure that the file path you are providing is correct with respect to the directory (folder) you are in, e.g. if you are referring simply to conf.txt in the command line, make sure you are in the same directory (folder) as this file. You can use ls or dir commands on Linux/MacOS and Windows, respectively, to list the contents of your directory.
Why do I get “usage errors” when I try to follow the video tutorial?
The command line options changed somewhat since the tutorial has been recorded. In particular, “--out” replaced “--all“.
Vina runs well on my machine, but when I run it on my exotic Linux cluster, I get a “boost thread resource” error. Why?
Your Linux cluster is [inadvertantly] configured in such a way as to disallow spawning threads. Therefore, Vina can not run. Contact your system administrator.
Why is my docked conformation different from what you get in the video tutorial?
The docking algorithm is non-deterministic. Even though with this receptor-ligand pair, the minimum of the scoring function corresponds to the correct conformation, the docking algorithm sometimes fails to find it. Try several times and see for yourself. Note that the probability of failing to find the mininum may be different with a different system.
My docked conformation is the same, but my energies are different from what you get in the video tutorial. Why?
The scoring function has changed since the tutorial was recorded, but only in the part that is independent of the conformation: the ligand-specific penalty for flexibility has changed.
Why do my results look weird in PyMOL?
PDBQT is not a standard molecular structure format. The version of PyMOL used in the tutorial (0.99rc6) happens to display it well (because PDBQT is somewhat similar to PDB). This might not be the case for newer versions of PyMOL.
Any other way to view the results?
You can also view PDBQT files in PMV (part of MGL Tools), or convert them into a different file format (e.g. using AutoDock Tools, or with “save as” in PMV)
How big should the search space be?
As small as possible, but not smaller. The smaller the search space, the easier it is for the docking algorithm to explore it. On the other hand, it will not explore ligand and flexible side chain atom positions outside the search space. You should probably avoid search spaces bigger than 30 x 30 x 30 Angstrom, unless you also increase “--exhaustiveness“.
Why am I seeing a warning about the search space volume being over 27000 Angstrom^3?
This is probably because you intended to specify the search space sizes in “grid points” (0.375 Angstrom), as in AutoDock 4. The AutoDock Vina search space sizes are given in Angstroms instead. If you really intended to use an unusually large search space, you can ignore this warning, but note that the search algorithm’s job may be harder. You may need to increase the value of the exhaustiveness to make up for it. This will lead to longer run time.
The bound conformation looks reasonable, except for the hydrogens. Why?
AutoDock Vina actually uses a united-atom scoring function, i.e. one that involves only the heavy atoms. Therefore, the positions of the hydrogens in the output are arbitrary. The hydrogens in the input file are used to decide which atoms can be hydrogen bond donors or acceptors though, so the correct protonation of the input structures is still important.
What does “exhaustiveness” really control, under the hood?
In the current implementation, the docking calculation consists of a number of independent runs, starting from random conformations. Each of these runs consists of a number of sequential steps. Each step involves a random perturbation of the conformation followed by a local optimization (using the Broyden-Fletcher-Goldfarb-Shanno algorithm) and a selection in which the step is either accepted or not. Each local optimization involves many evaluations of the scoring function as well as its derivatives in the position-orientation-torsions coordinates. The number of evaluations in a local optimization is guided by convergence and other criteria. The number of steps in a run is determined heuristically, depending on the size and flexibility of the ligand and the flexible side chains. However, the number of runs is set by the exhaustiveness parameter. Since the individual runs are executed in parallel, where appropriate, exhaustiveness also limits the parallelism. Unlike in AutoDock 4, in AutoDock Vina, each run can produce several results: promising intermediate results are remembered. These are merged, refined, clustered and sorted automatically to produce the final result.
Why do I not get the correct bound conformation?
It can be any of a number of things:
- If you are coming from AutoDock 4, a very common mistake is to specify the search space in “points” (0.375 Angstrom), instead of Angstroms.
- Your ligand or receptor might not have been correctly protonated.
- Bad luck (the search algorithm could have found the correct conformation with good probability, but was simply unlucky). Try again with a different seed.
- The minimum of the scoring function correponds to the correct conformation, but the search algorithm has trouble finding it. In this case, higher exhaustiveness or smaller search space should help.
- The minimum of the scoring function simply is not where the correct conformation is. Trying over and over again will not help, but may occasionally give the right answer if two wrongs (inexact search and scoring) make a right. Docking is an approximate approach.
- Related to the above, the culprit may also be the quality of the X-ray or NMR receptor structure.
- If you are not doing redocking, i.e. using the correct induced fit shape of the receptor, perhaps the induced fit effects are large enough to affect the outcome of the docking experiment.
- The rings can only be rigid during docking. Perhaps they have the wrong conformation, affecting the outcome.
- You are using a 2D (flat) ligand as input.
- The actual bound conformation of the ligand may occasionally be different from what the X-ray or NMR structure shows.
- Other problems
How can I tweak the scoring function?
You can change the weights easily, by specifying them in the configuration file, or in the command line. For example
vina --weight_hydrogen -1.2 ...
doubles the strenth of all hydrogen bonds.
Functionality that would allow the users to create new atom and pseudo-atom types, and specify their own interaction functions is planned for the future.
This should make it easier to adapt the scoring function to specific targets, model covalent docking and macro-cycle flexibility, experiment with new scoring functions, and, using pseudo-atoms, create directional interaction models.
Stay tuned to the AutoDock mailing list, if you wish to be notified of any beta-test releases.
Why don’t I get as many binding modes as I specify with “--num_modes“?
This option specifies the maximum number of binding modes to output. The docking algorithm may find fewer “interesting” binding modes internally. The number of binding modes in the output is also limited by the “energy_range“, which you may want to increase.
Why don’t the results change when I change the partial charges?
AutoDock Vina ignores the user-supplied partial charges. It has its own way of dealing with the electrostatic interactions through the hydrophobic and the hydrogen bonding terms. See the original publication [*] for details of the scoring function.
I changed something, and now the docking results are different. Why?
Firstly, had you not changed anything, some results could have been different anyway, due to the non-deterministic nature of the search algorithm. Exact reproducibility can be assured by supplying the same random seed to both calculations, but only if all other inputs and parameters are the same as well. Even minor changes to the input can have an effect similar to a new random seed. What does make sense discussing are the statistical properties of the calculations: e.g. “with the new protonation state, Vina is much less likely to find the correct docked conformation”.
How do I use flexible side chains?
You split the receptor into two parts: rigid and flexible, with the latter represented somewhat similarly to how the ligand is represented. See the section “Flexible Receptor PDBQT Files” of the AutoDock4.2 User Guide (page 14) for how to do this in AutoDock Tools. Then, you can issue this command: vina --config conf --receptor rigid.pdbqt --flex side_chains.pdbqt --ligand ligand.pdbqt. Also see this write-up on this subject.
How do I do virtual screening?
Please see the relevant section of the manual.
Please note that a variety of docking management applications exist to assist you in this task.
I don’t have sufficient computing resources to run a virtual screen. What are my options?
You may be able to run your project on the World Community Grid, or use DrugDiscovery@TACC. See Other Software.
I have ideas for new features and other suggestions.
For proposed new features, we like there to be a wide consensus, resulting from a public discussion, regarding their necessity. Please consider starting or joining a discussion on the AutoDock mailing list.
Will you answer my questions about Vina if I email or call you?
No. Vina is community-supported. There is no obligation on the authors to help others with their projects. Please see this page for how to get help.
Platform Notes and Installation
Windows
Compatibility
Vina is expected to work on Windows XP and newer systems.
Installing
Double-click the downloaded MSI file and follow the instructions
Running
Open the Command Prompt and, if you installed Vina in the default location, type
"\Program Files\The Scripps Research Institute\Vina\vina.exe" --help
If you are using Cygwin, the above command would instead be
/cygdrive/c/Program\ Files/The\ Scripps\ Research\ Institute/Vina/vina --help
See the Video Tutorial for details. Don’t forget to check out Other Software for GUIs, etc.
Linux
Compatibility
Vina is expected to work on x86 and compatible 64-bit Linux systems.
Installing
tar xzvf autodock_vina_1_1_2_linux_x86.tgz
Optionally, you can copy the binary files where you want.
Running
./autodock_vina_1_1_2_linux_x86/bin/vina --help
If the executable is in your PATH, you can just type “vina --help” instead. See the Video Tutorial for details. Don’t forget to check out Other Software for GUIs, etc.
Mac
Compatibility
The 64 bit version is expected to work on Mac OS X 10.15 (Catalina) and newer. The 32 bit version of Vina is expected to work on Mac OS X from 10.4 (Tiger) through 10.14 (Mojave).
Installing
tar xzvf autodock_vina_1_1_2_mac_64bit.tgz # 64 bit tar xzvf autodock_vina_1_1_2_mac.tgz # 32 bit
Optionally, you can copy the binary files where you want.
Running
./autodock_vina_1_1_2_mac_64bit/bin/vina --help # 64 bit ./autodock_vina_1_1_2_mac/bin/vina --help # 32 bit
If the executable is in your PATH, you can just type “vina --help” instead. See the Video Tutorial for details. Don’t forget to check out Other Software for GUIs, etc.
Building from Source
Attention: Building Vina from source is NOT meant to be done by regular users! (these instructions might be outdated)
Step 1: Install a C++ compiler suite
On Windows, you may want to install Visual Studio; on OS X, Xcode; and on Linux, the GCC compiler suite.
Step 2: Install Boost
Install Boost. (Version 1.41.0 was used to compile the official binaries. With other versions, your luck may vary) Then, build and run one of the example programs, such as the Regex example, to confirm that you have completed this step. If you can’t do this, please seek help from the Boost community.
Step 3: Build Vina
If you are using Visual Studio, you may want to create three projects: lib, main and split, with the source code from the appropriate subdirectories. lib must be a library, that the other projects depend on, and main and split must be console applications. For optimal performance, remember to compile using the Release mode.
On OS X and Linux, you may want to navigate to the appropriate build subdirectory, customize the Makefile by setting the paths and the Boost version, and then type
make depend make
Other Software
Disclaimer: This list is for information purposes only and does not constitute an endorsement.
- Tools specifically designed for use with AutoDock Vina (in no particular order):
- MGLTools, which includes AutoDock Tools (ADT) and Python Molecular Viewer (PMV). ADT is required for generating input files for AutoDock Vina, and PMV can be used for viewing the results
- PyRx can be used to set up docking and virtual screening with AutoDock Vina and to view the results
- The new Autodock/Vina plugin for PyMOL can be used to set up docking and virtual screening with AutoDock Vina and to view the results
- Computer-Aided Drug-Design Platform using PyMOL is another plugin for PyMOL that also integrates AMBER, Reduce and SLIDE.
- AutoGrow uses AutoDock Vina in its rational drug design procedure
- NNScore will re-score Vina results using an artificial neural network trained on Binding MOAD and PDBBind
- A Vina GUI layer for Windows by Biochem Lab Solutions can be used to facilitate virtual screening with AutoDock Vina
- VSDK can be used to faciliate virtual screening with AutoDock Vina
- PaDEL-ADV can be used to facilitate virtual screening with AutoDock Vina
- DrugDiscovery@TACC allows you to do virtual screening with AutoDock Vina through their web site
- NBCR CADD Pipeline provides access to virtual screening with AutoDock Vina on NBCR computers
- MOLA is a bootable, self-configuring system for virtual screening using AutoDock4/Vina on computer clusters
- SMINA is a modification of Vina that links with OpenBabel for I/O and supports additional tweaks of the scoring function
- Off-Target Pipeline is a platform intended to carry out secondary target identification and docking
- AUDocker can be used to facilitate virtual screening with AutoDock Vina
- World Community Grid can be used by qualified projects to run AutoDock Vina calculations for free on the massively parallel network of computers, where volunteers donate their idle CPU time.
- DockoMatic is a graphical user interface intended to facilitate virtual screening with AutoDock and AutoDock Vina.
- VinaLC is a modification of Vina by the Lawrence Livermore National Laboratory that takes advantage of MPI on computer clusters
- Other tools that you are likely to find useful while docking or virtual screening with AutoDock Vina:
- PyMOL is one of the most popular programs for molecular visualization and can be used for viewing the docking results
- OpenBabel can be used to convert among various structure file formats, assign the protonation states, etc.
- ChemAxon Marvin can be used to visualize structures, convert among various structure file formats, assign the protonation states, etc.
Usage
Summary
The usage summary can be obtained with “vina --help“:
Input:
--receptor arg rigid part of the receptor (PDBQT)
--flex arg flexible side chains, if any (PDBQT)
--ligand arg ligand (PDBQT)
Search space (required):
--center_x arg X coordinate of the center
--center_y arg Y coordinate of the center
--center_z arg Z coordinate of the center
--size_x arg size in the X dimension (Angstroms)
--size_y arg size in the Y dimension (Angstroms)
--size_z arg size in the Z dimension (Angstroms)
Output (optional):
--out arg output models (PDBQT), the default is chosen based on
the ligand file name
--log arg optionally, write log file
Misc (optional):
--cpu arg the number of CPUs to use (the default is to try to
detect the number of CPUs or, failing that, use 1)
--seed arg explicit random seed
--exhaustiveness arg (=8) exhaustiveness of the global search (roughly
proportional to time): 1+
--num_modes arg (=9) maximum number of binding modes to generate
--energy_range arg (=3) maximum energy difference between the best binding
mode and the worst one displayed (kcal/mol)
Configuration file (optional):
--config arg the above options can be put here
Information (optional):
--help display usage summary
--help_advanced display usage summary with advanced options
--version display program version
Configuration File
For convenience, some command line options can be placed into a configuration file.
For example:
receptor = hsg1/rigid.pdbqt ligand = ligand.pdbqt center_x = 2 center_y = 6 center_z = -7 size_x = 25 size_y = 25 size_z = 25 energy_range = 4
In case of a conflict, the command line option takes precedence over the configuration file one.
Search space
The search space effectively restricts where the movable atoms, including those in the flexible side chains, should lie.
Exhaustiveness
With the default (or any given) setting of exhaustiveness, the time spent on the search is already varied heuristically depending on the number of atoms, flexibility, etc. Normally, it does not make sense to spend extra time searching to reduce the probability of not finding the global minimum of the scoring function beyond what is significantly lower than the probability that the minimum is far from the native conformation. However, if you feel that the automatic trade-off made between exhaustiveness and time is inadequate, you can increase the exhaustiveness level. This should increase the time linearly and decrease the probability of not finding the minimum exponentially.
Output
Energy
The predicted binding affinity is in kcal/mol.
RMSD
RMSD values are calculated relative to the best mode and use only movable heavy atoms. Two variants of RMSD metrics are provided, rmsd/lb (RMSD lower bound) and rmsd/ub (RMSD upper bound), differing in how the atoms are matched in the distance calculation:
rmsd/ubmatches each atom in one conformation with itself in the other conformation, ignoring any symmetryrmsd'matches each atom in one conformation with the closest atom of the same element type in the other conformation (rmsd'can not be used directly, because it is not symmetric)rmsd/lbis defined as follows:rmsd/lb(c1, c2) = max(rmsd'(c1, c2), rmsd'(c2, c1))
Hydrogen positions
Vina uses a united-atom scoring function. As in AutoDock, polar hydrogens are needed in the input structures to correctly type heavy atoms as hydrogen bond donors. However, in Vina, the degrees of freedom that only move hydrogens, such as the hydroxyl group torsions, are degenerate. Therefore, in the output, some hydrogen atoms can be expected to be positioned randomly (but consistent with the covalent structure). For a united-atom treatment, this is essentially a cosmetic issue.
Separate models
All predicted binding modes, including the positions of the flexible side chains are placed into one multimodel PDBQT file specified by the “out” parameter or chosen by default, based on the ligand file name. If needed, this file can be split into individual models using a separate program called “vina_split”, included in the distribution.
Advanced Options
AutoDock Vina’s “advanced options” are intended to be primarily used by people interested in methods development rather than the end users. The usage summary including the advanced options can be shown with
vina --help_advanced
The advanced options allow
- scoring without minimization
- performing local optimization only
- randomizing the input with no search (this is useful for testing docking software)
- changing the weights from their default values (see the paper[*] for what the weights mean)
- displaying the individual contributions to the intermolecular score, before weighting (these are shown with “
--score_only“; see the paper[2] for what the terms are)
Virtual Screening
You may want to choose some of the tools listed under Other Software to perform virtual screening. Alternatively, if you are familiar with shell scripting, you can do virtual screening without them.
The examples below assume that Bash is your shell. They will need to be adapted to your specific needs.
Windows
To perform virtual screening on Windows, you can either use Cygwin and the Bash scripts below, or, alternatively, adapt them for the Windows scripting language.
Linux, Mac
Suppose you are in a directory containing your receptor receptor.pdbqt and a set of ligands named ligand_01.pdbqt, ligand_02.pdbqt, etc.
You can create a configuration file conf.txt, such as
receptor = receptor.pdbqt center_x = 2 center_y = 6 center_z = -7 size_x = 25 size_y = 25 size_z = 25 num_modes = 9
and dock all ligands with this shell script. The script assumes that vina is in your PATH. Otherwise, modify it accordingly.
PBS Cluster
If you have a Linux Beowulf cluster, you can perform the individual dockings in parallel.
Continuing with our example, instead of executing all the dockings in a loop locally, we will write one *.job script per ligand, and use qsub (a PBS command) to schedule these scripts to be executed by the cluster.
Run this shell script to do it. The script assumes that vina and qsub are in your PATH. Otherwise, modify it accordingly.
Once the jobs have been scheduled, you can monitor their status with
qstat -u `whoami`
Selecting Best Results
If you are on Unix and in a directory that contains directories with PDBQT files, all of which are AutoDock Vina results, you may find this Python script useful for selecting the top results. Run it as:
vina_screen_get_top.py 10
to get the file names of the top 10 hits, which can then be easily copied.
History
Brief summaries of changes between versions can be found here.
Citation
If you used AutoDock Vina in your work, please cite:
Getting Help
Please see this page if you have questions about AutoDock Vina.
Reporting Bugs
Potential bug reports are greatly appreciated, even if you are not exactly sure that they are bugs. However, please do not include requests for assistance along with your bug report. See this page instead.
Likely bugs:
- Early termination
- Failure to terminate
- Changes of the covalent lengths or of the invariant angles in the output
- “Obviously wrong” clashes (check your “search space” though)
- Disagreement with the documentation
Likely not bugs:
- Anything that happens before you run Vina or after it finished
- Occasional disagreement with the experiment
- Vina’s refusal to open a file that does not exist (e.g. try
ls conf.txtto see if the file is really there)
Reporting
You can send your reports to the AutoDock mailing list. Please remember to provide a descriptive “Subject” line and all of the information needed to reproduce the problem you are seeing.